*AIMOVIG® IS PCRS REIMBURSABLE FOR CHRONIC MIGRAINE PATIENTS WITH THREE OR MORE PREVIOUS PROPHYLACTIC TREATMENT FAILURES.
ABBREVIATED PRESCRIBING INFORMATION
▼ Aimovig® (erenumab) 70 mg and 140 mg solution for injection in pre-filled pen
Important note: Before prescribing, consult full prescribing information.
Presentation: Aimovig 70 mg solution for injection in pre-filled pen. Each pre-filled pen contains 70 mg erenumab. Aimovig 140 mg solution for injection in pre-filled pen. Each pre-filled pen contains 140 mg erenumab.
Indications: Aimovig is indicated for prophylaxis of migraine in adults who have at least 4 migraine days per month.
Dosage and administration: Adults: The recommended dose of Aimovig is 70 mg administered subcutaneously every 4 weeks. Some patients may benefit from a dosage of 140 mg once every 4 weeks. Each 140 mg dose is given either as one subcutaneous injection of 140 mg or as two subcutaneous injections of
70 mg. Aimovig is intended for patient self-administration in the abdomen, thigh, or, if someone else is giving the injection, also into the outer area of the upper arm. Administration should be performed by an individual who has been trained to administer the product. The needle cover of Aimovig prefilled pen contains dry natural rubber, which may cause allergic reactions in individuals sensitive to latex.
Special populations: Paediatric patients: The safety and effectiveness of Aimovig has not been studied in paediatric patients.
Elderly (aged 65 years and over): The safety and effectiveness of Aimovig has not been studied in elderly patients. No dose adjustment is necessary as the pharmacokinetics of erenumab are not affected by age.
Renal impairment: No dose adjustment is necessary in patients with mild to moderate renal impairment.
Hepatic impairment: No studies have been performed in patients with hepatic impairment. Hepatic clearance is not a major clearance pathway for erenumab.
Contraindications: Hypersensitivity to the active substance or to any of the excipients.
Warnings and precautions: Hypersensitivity reactions: Serious hypersensitivity reactions, including rash, angioedema, and anaphylactic reactions, have been reported with erenumab in post-marketing experience. These reactions may occur within minutes, although some may occur more than one week after treatment. In that context, patients should be warned about the symptoms associated with hypersensitivity reactions. If a serious or severe hypersensitivity reaction occurs, initiate appropriate therapy and do not continue treatment with erenumab. Constipation: Constipation is a common undesirable effect of Aimovig and is usually mild or moderate in intensity. In a majority of the cases, the onset was reported after the first dose of Aimovig; however patients have also experienced constipation later on in the treatment. In most cases constipation resolved within three months. In the post marketing setting, constipation with serious complications has been reported with erenumab. In some of these cases hospitalisation was required, including cases where surgery was necessary. History of constipation or the concurrent use of medicinal products associated with decreased gastrointestinal motility may increase the risk for more severe constipation and the potential for constipation related complications. Patients should be warned about the risk of constipation and advised to seek medical attention in case constipation does not resolve or worsens. Patients should seek medical attention immediately if they develop severe constipation. Constipation should be managed promptly as clinically appropriate. For severe constipation, discontinuation of treatment should be considered. Traceability: In order to improve the traceability of biological medicinal products, the name and the batch number of the administered product should be clearly recorded. Latex sensitive individuals: The removable cap of the Aimovig pre-filled pen contains dry natural rubber latex, which may cause Allergic reactions in individuals sensitive to latex Pregnancy, lactation, fertility: Pregnancy: There are a limited amount of data from the use of erenumab in pregnant women. As a precautionary measure it is preferable to avoid the use of Aimovig during pregnancy. Lactation: It is not known whether erenumab is present in human milk. The use of Aimovig could be considered during breastfeeding only if clinically needed. Fertility: There is no data available on the impact of Aimovig on male and female fertility. Animal studies showed no impact on female and male fertility.
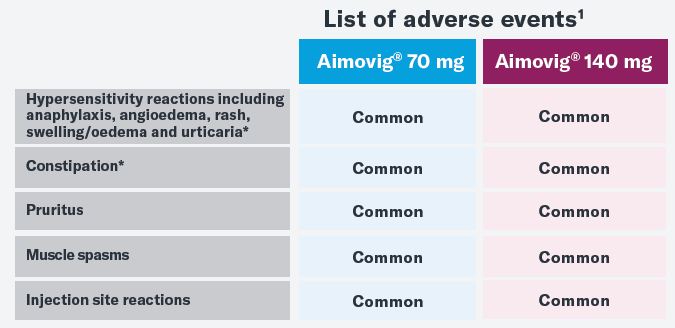
Adverse drug reactions: Common (≥1/100 to <1/10): Hypersensitivity reactions including anaphylaxis, angioedema, rash, swelling/oedema and urticaria. Injection site reactions, constipation, muscle spasm, pruritus.
Interactions: No effect on exposure of co-administered medicinal products is expected based on the metabolic pathway of monoclonal antibodies. No interactions with oral contraceptives (ethinyl estradiol and norgestimate) or sumatriptan were observed in studies with healthy volunteers.
Legal Category: POM.
Marketing Authorisation Holder: Novartis Europharm Ltd, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland.
Marketing Authorisation Numbers: EU/1/18/1293/001 006.
Date of last revision of Abbreviated Prescribing Information: September 2020.
Full prescribing information is available upon request from: Novartis Ireland Limited, Vista Building, Elm Park Business Campus, Merrion
Road, Dublin 4, Ireland, Tel: + 353 1 220 4100 or at
www.medicines.ie. Detailed information on this product is also available on the website of the European Medicines Agency http://www.ema.europa.eu.
▼ This medicinal product is subject to additional monitoring. Reporting suspected adverse reactions of the medicinal product is important to Novartis and the HPRA. It allows continued monitoring of the benefit/risk profile of the medicinal product. All suspected adverse reactions should be reported via HPRA Pharmacovigilance, website: www.hpra.ie. Adverse events could also be reported to Novartis preferably via www.report.novartis.com or by email: [email protected] or by calling
01 2080 612.